Create a genome-wide Manhattan plot from GWAS summary statistics. Returns a ggplot2 object for further customization.
Usage
manhattan_plot(
data,
chr = NULL,
bp = NULL,
p = NULL,
snp = NULL,
colors = c("#1A5276", "#76D7C4"),
point_size = 0.8,
alpha = 1,
genome_wide = 5e-08,
suggestive = 1e-05,
threshold_colors = c("#E74C3C", "#3498DB"),
highlight_snps = NULL,
highlight_color = "#E74C3C",
highlight_size = 2,
label_snps = NULL,
label_top_n = NULL,
label_column = "SNP",
downsample = TRUE,
downsample_n = 2e+05,
chromosomes = NULL,
chr_labels = NULL,
y_metric = "p",
y_limit = NULL,
y_truncate = NULL,
title = NULL
)Arguments
- data
A
gwas_dataobject or data.frame with GWAS results.- chr, bp, p, snp
Column name overrides (auto-detected if NULL).
- colors
Two-color vector for alternating chromosomes.
- point_size
Point size.
- alpha
Point transparency.
- genome_wide
Genome-wide significance threshold (p-value).
- suggestive
Suggestive significance threshold.
- threshold_colors
Colors for significance lines.
- highlight_snps
Character vector of SNP IDs to highlight.
- highlight_color
Color for highlighted SNPs.
- highlight_size
Size for highlighted points.
- label_snps
Character vector of SNP IDs to label.
- label_top_n
Label the top N most significant SNPs.
- label_column
Column to use for label text.
- downsample
Enable smart downsampling for large datasets.
- downsample_n
Target number of points after downsampling.
- chromosomes
Subset of chromosomes to plot (integer vector).
- chr_labels
Custom chromosome labels. Options: NULL (all labels),
"odd"(only odd-numbered chromosomes labeled), or a character vector of labels (same length as displayed chromosomes).- y_metric
What to plot on the y-axis.
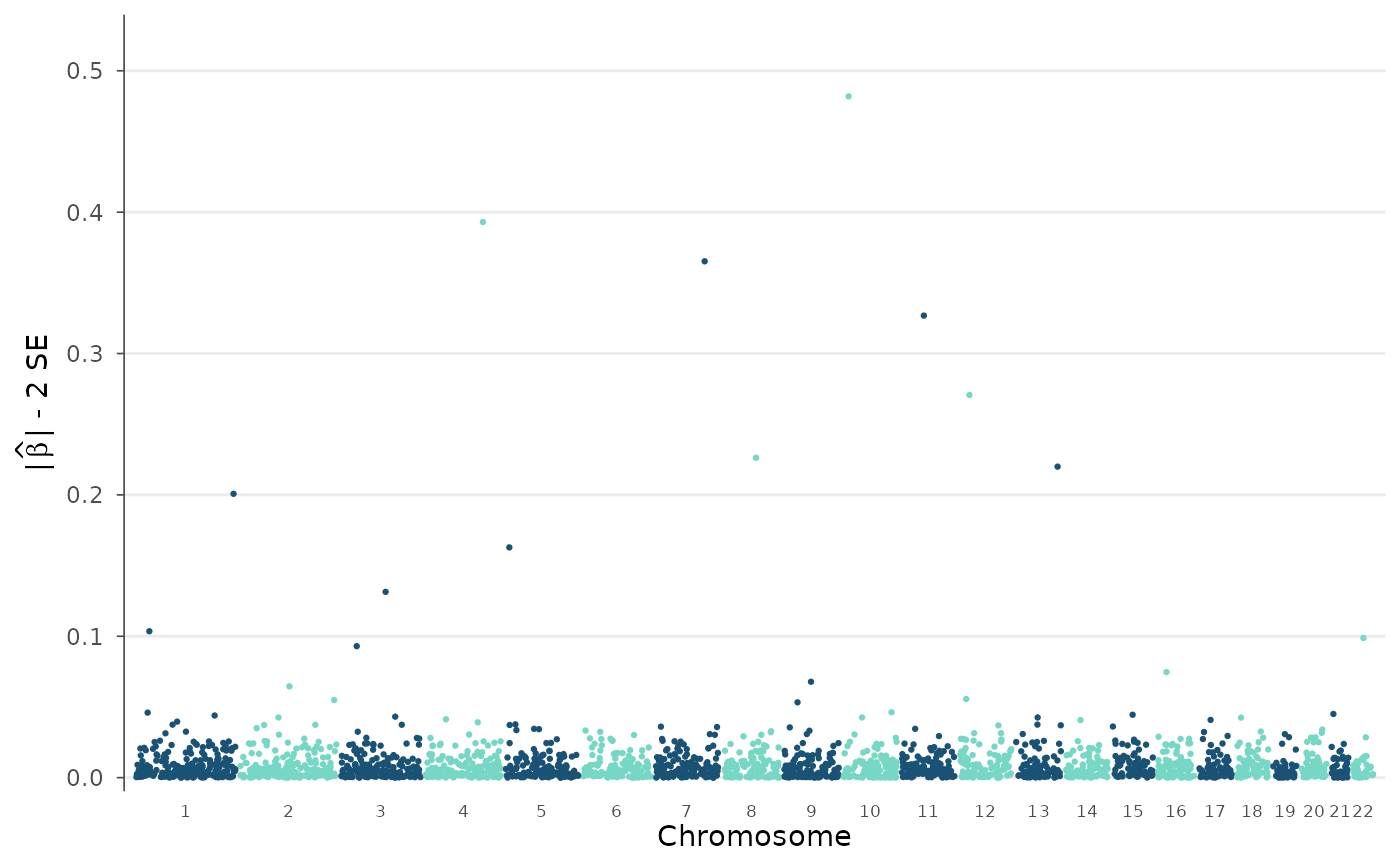
"p"(default) shows -log10(p)."beta_min"shows the lower confidence bound of the absolute effect size: |beta| - 2*SE. Variants whose confidence interval overlaps zero are excluded. This highlights variants with large, robust effect sizes rather than just small p-values.- y_limit
Upper y-axis limit for -log10(p).
- y_truncate
Break the y-axis for datasets with extreme peaks. A single value shows 0 to that value at full scale and compresses everything above it into a band above the break (nothing is dropped), e.g.
y_truncate = 15. A vector of two valuesc(break_from, resume_at)instead cuts out the middle region:y_truncate = c(15, 50)shows 0-15 at full scale, drops 15-50, then shows 50+ above the break. Units are -log10(p).- title
Plot title.
Examples
data(example_gwas, package = "ggwas")
# Basic Manhattan plot
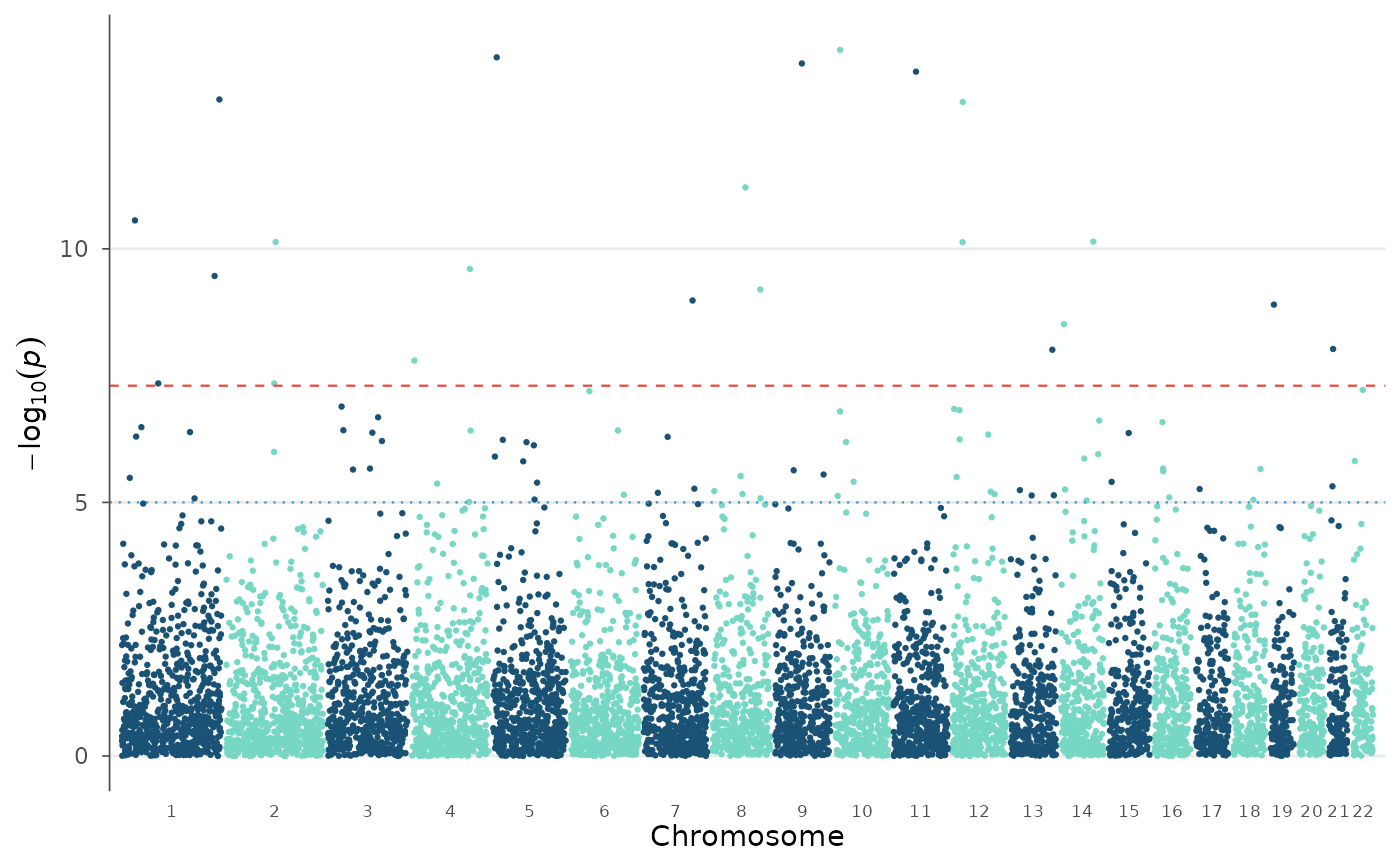
manhattan_plot(example_gwas)
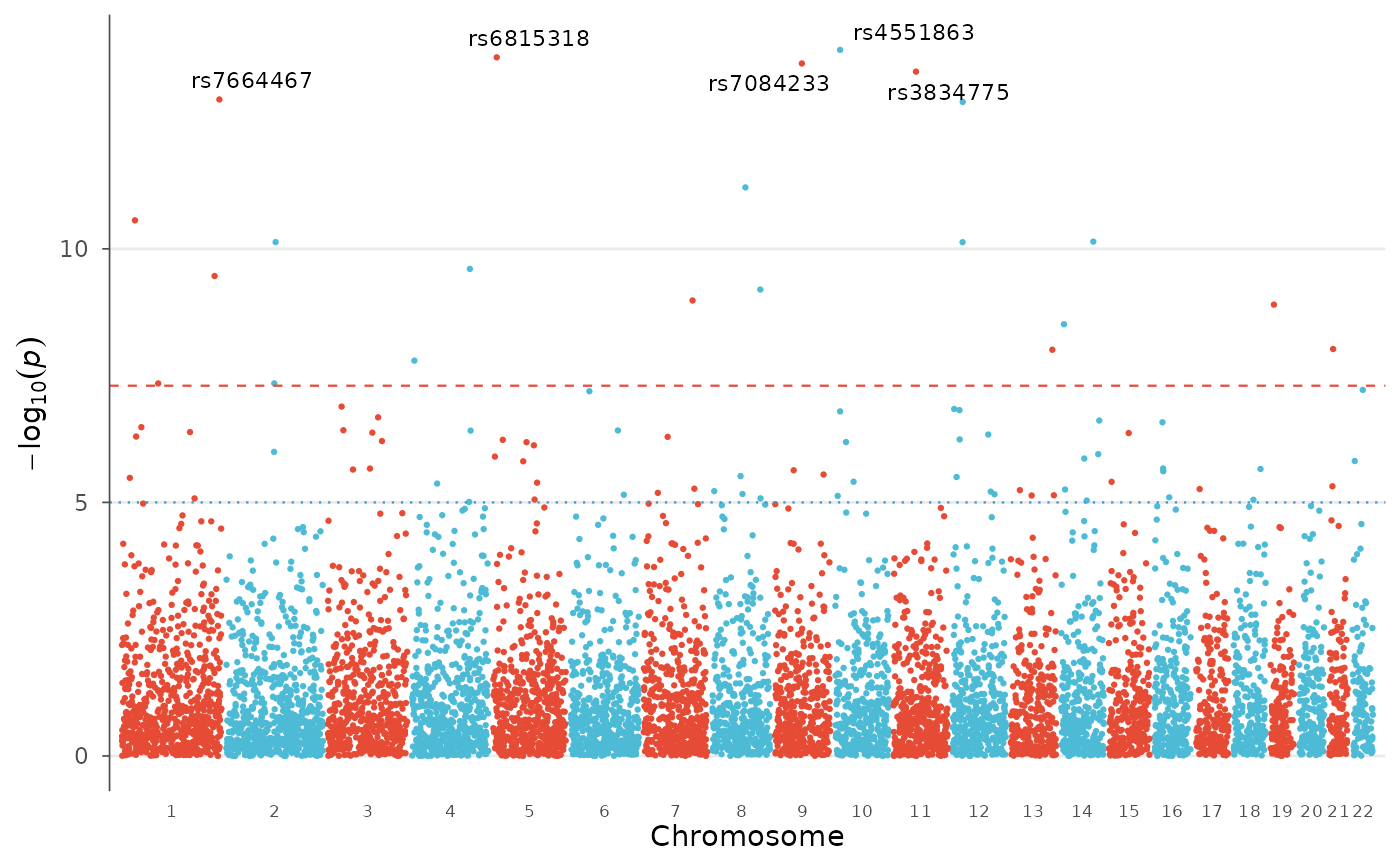
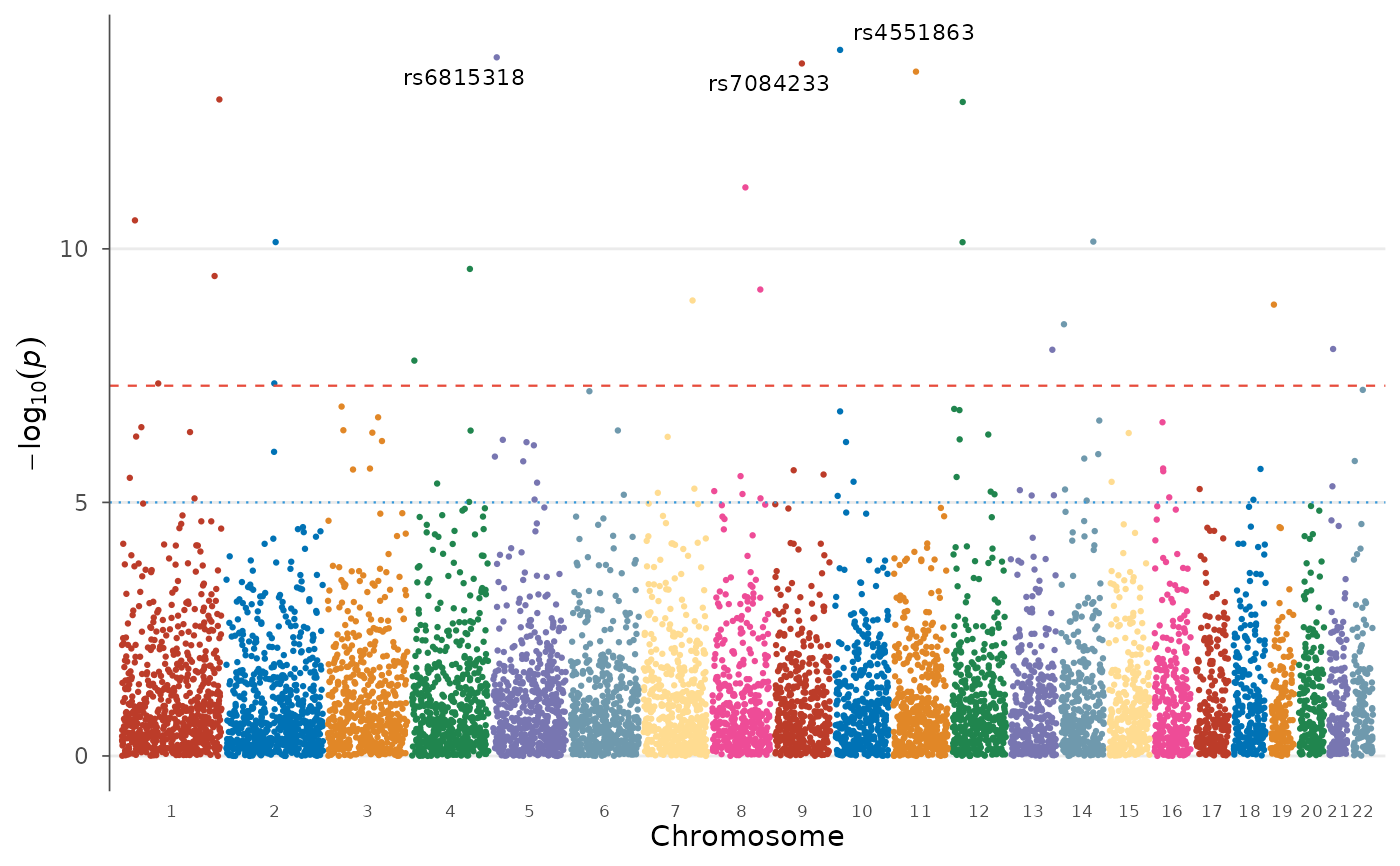
 # Label top hits with a different palette
manhattan_plot(example_gwas, label_top_n = 5, colors = gwas_palette("vibrant"))
# Label top hits with a different palette
manhattan_plot(example_gwas, label_top_n = 5, colors = gwas_palette("vibrant"))
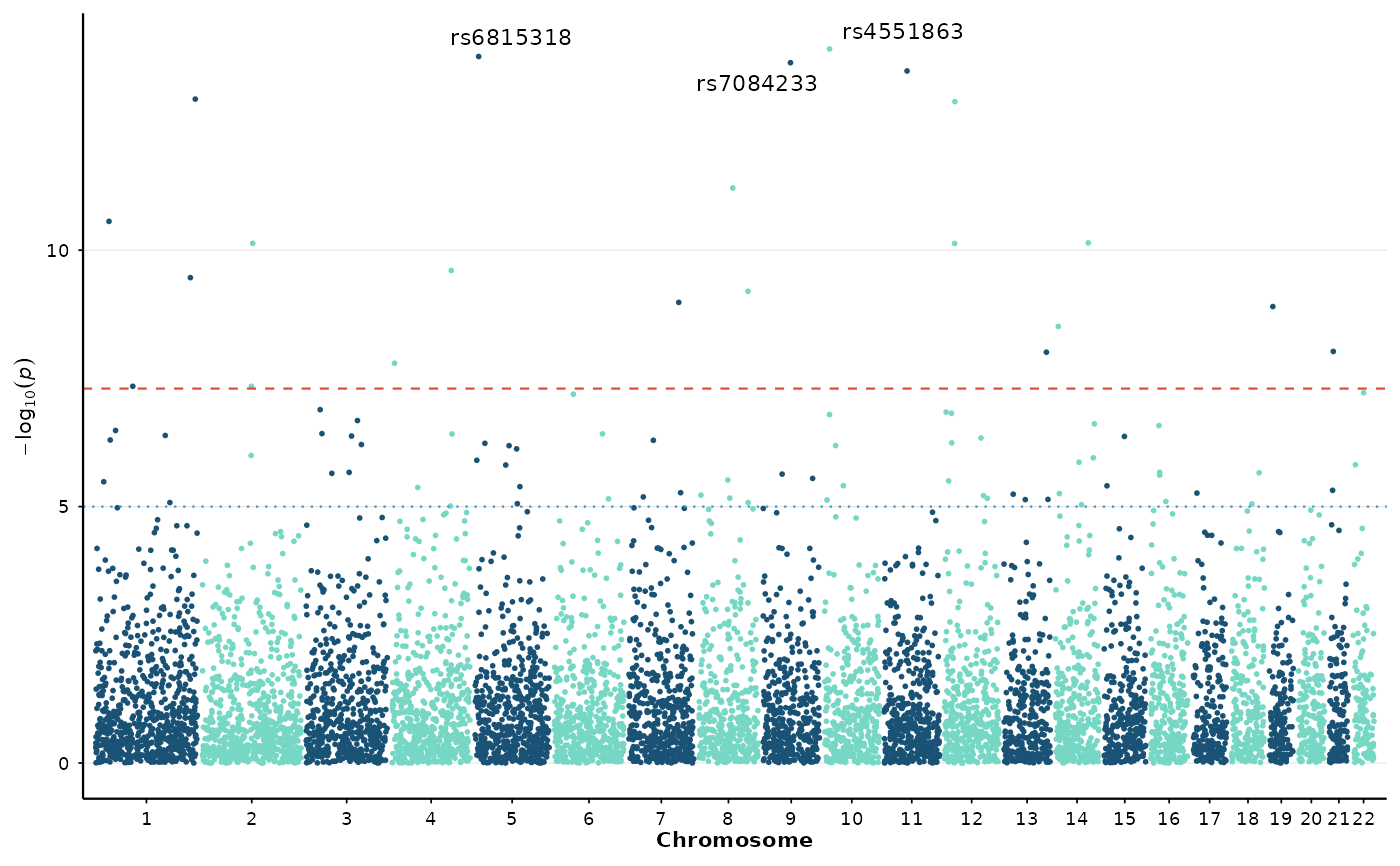
 # Nature journal style
manhattan_plot(example_gwas, label_top_n = 3) + theme_nature()
# Nature journal style
manhattan_plot(example_gwas, label_top_n = 3) + theme_nature()
 # Lancet palette with Science theme
manhattan_plot(example_gwas, colors = gwas_palette("lancet")) + theme_science()
# Lancet palette with Science theme
manhattan_plot(example_gwas, colors = gwas_palette("lancet")) + theme_science()
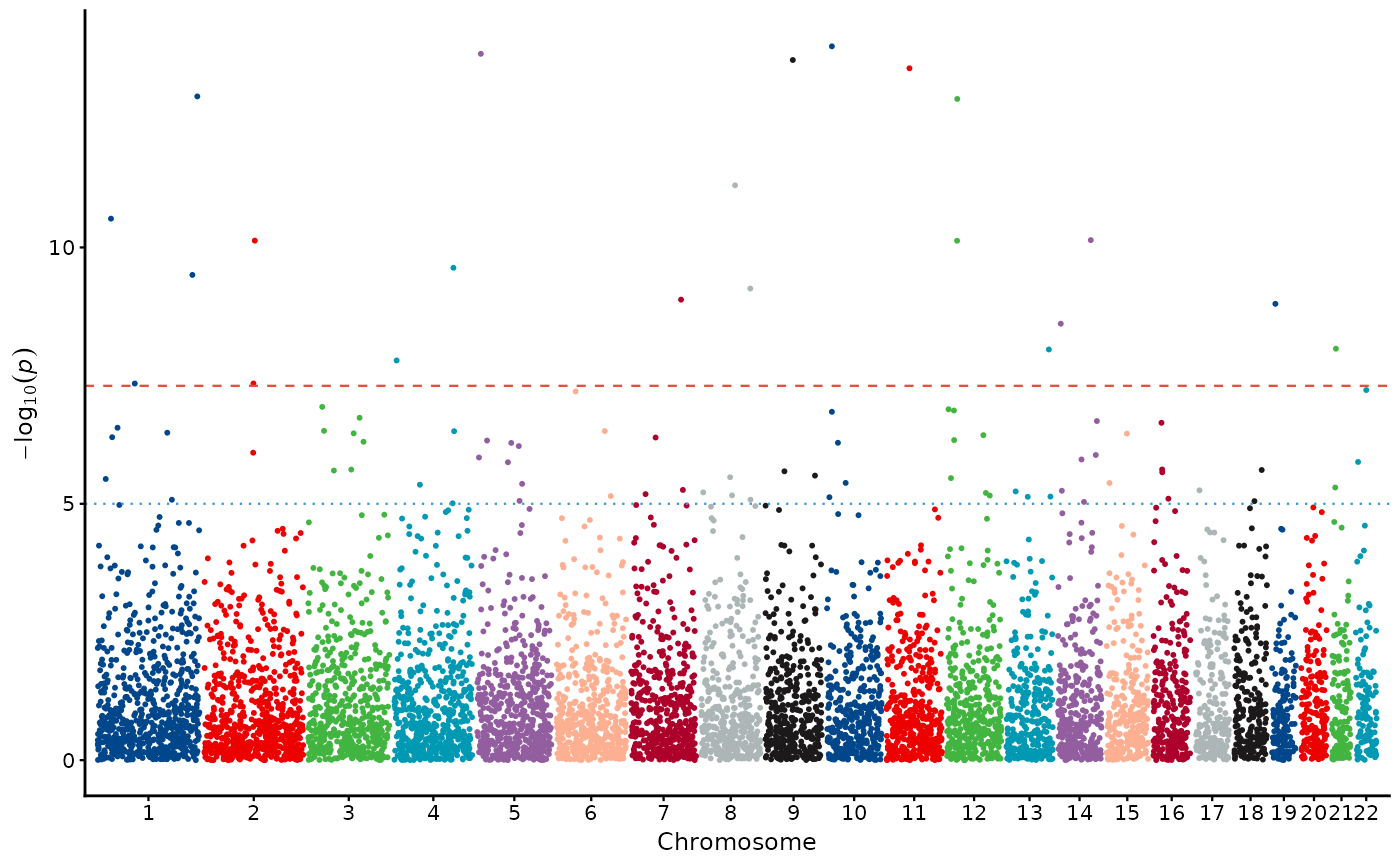 # Subset chromosomes
manhattan_plot(example_gwas, chromosomes = seq_len(10))
# Subset chromosomes
manhattan_plot(example_gwas, chromosomes = seq_len(10))
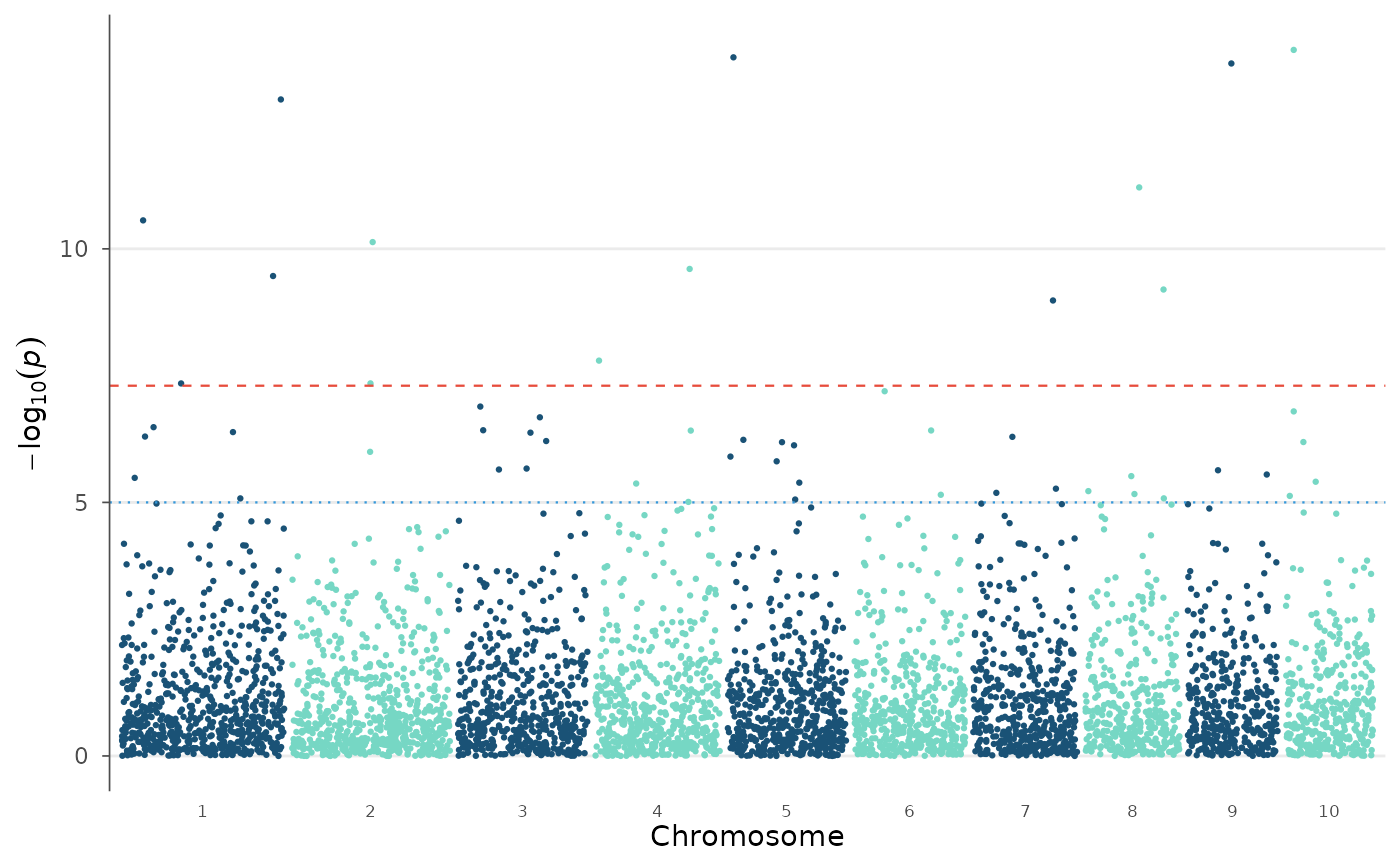 # NEJM palette
manhattan_plot(example_gwas, colors = gwas_palette("nejm"), label_top_n = 3)
# NEJM palette
manhattan_plot(example_gwas, colors = gwas_palette("nejm"), label_top_n = 3)
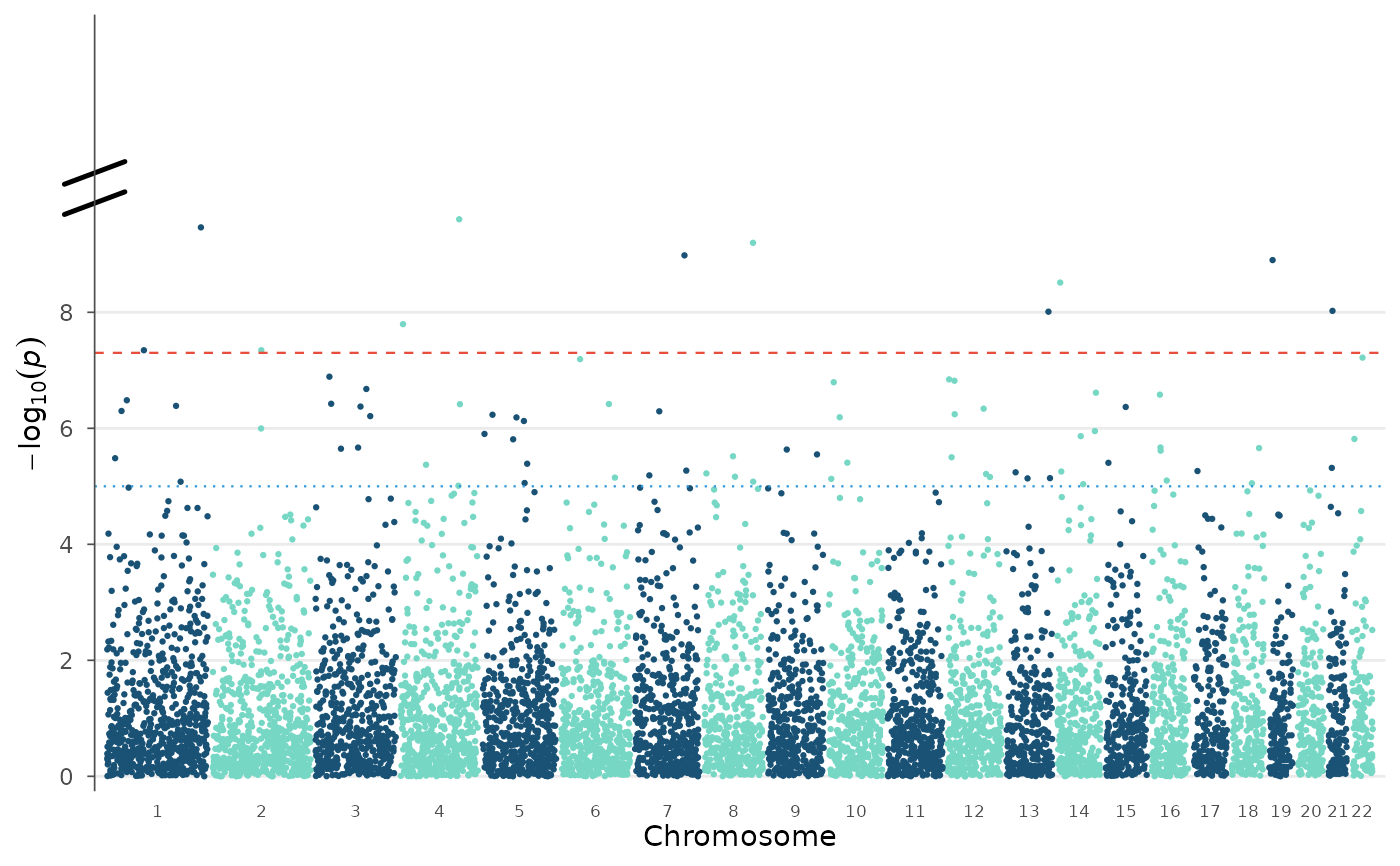
 # Broken y-axis: cut 10-50, show 0-10 and 50+
manhattan_plot(example_gwas, y_truncate = c(10, 50))
# Broken y-axis: cut 10-50, show 0-10 and 50+
manhattan_plot(example_gwas, y_truncate = c(10, 50))
 # Single value: auto-detect resume point
manhattan_plot(example_gwas, y_truncate = 10)
# Single value: auto-detect resume point
manhattan_plot(example_gwas, y_truncate = 10)
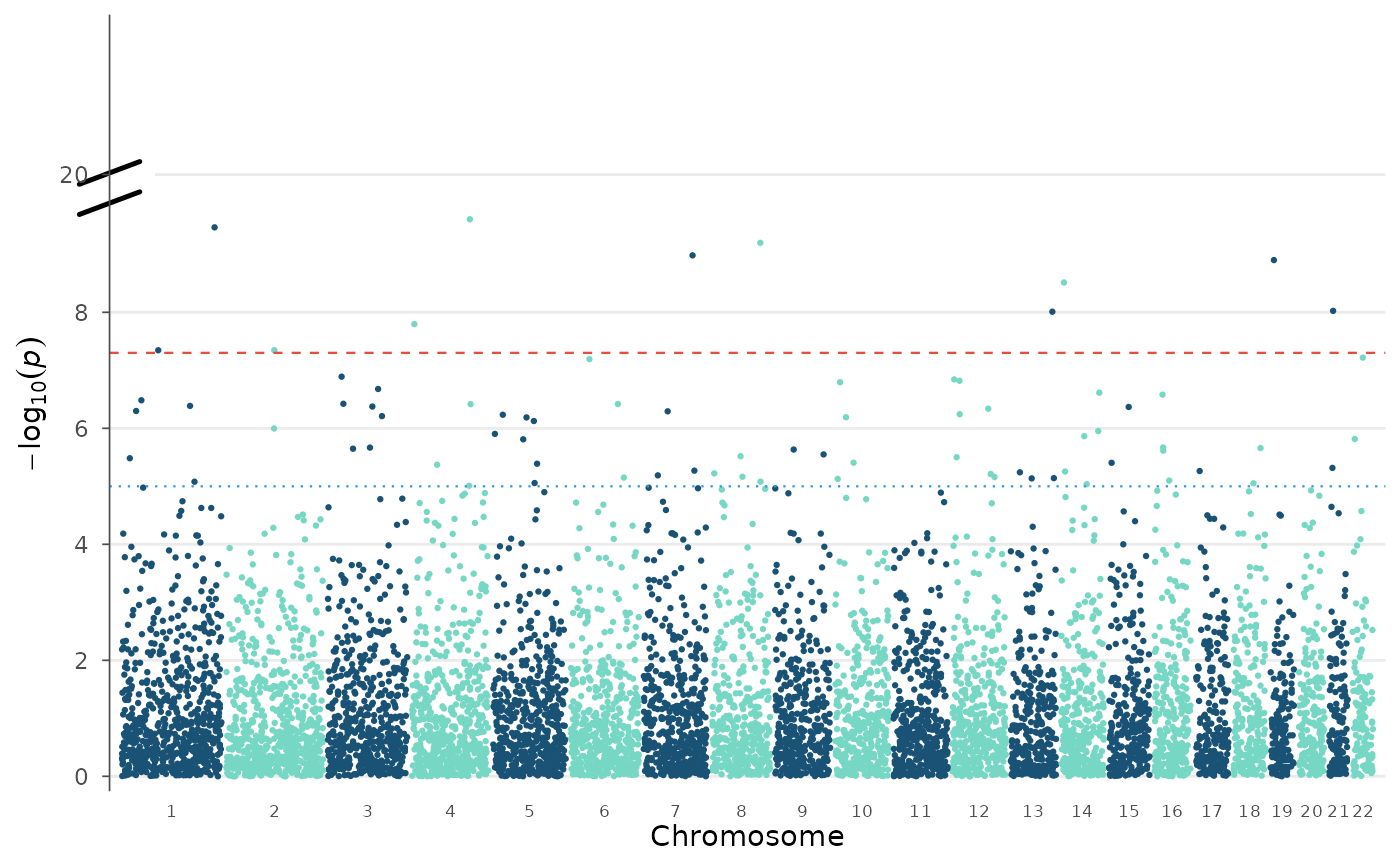 # Custom significance threshold (Bonferroni for 500k SNPs)
manhattan_plot(example_gwas, genome_wide = 0.05 / 500000)
# Custom significance threshold (Bonferroni for 500k SNPs)
manhattan_plot(example_gwas, genome_wide = 0.05 / 500000)
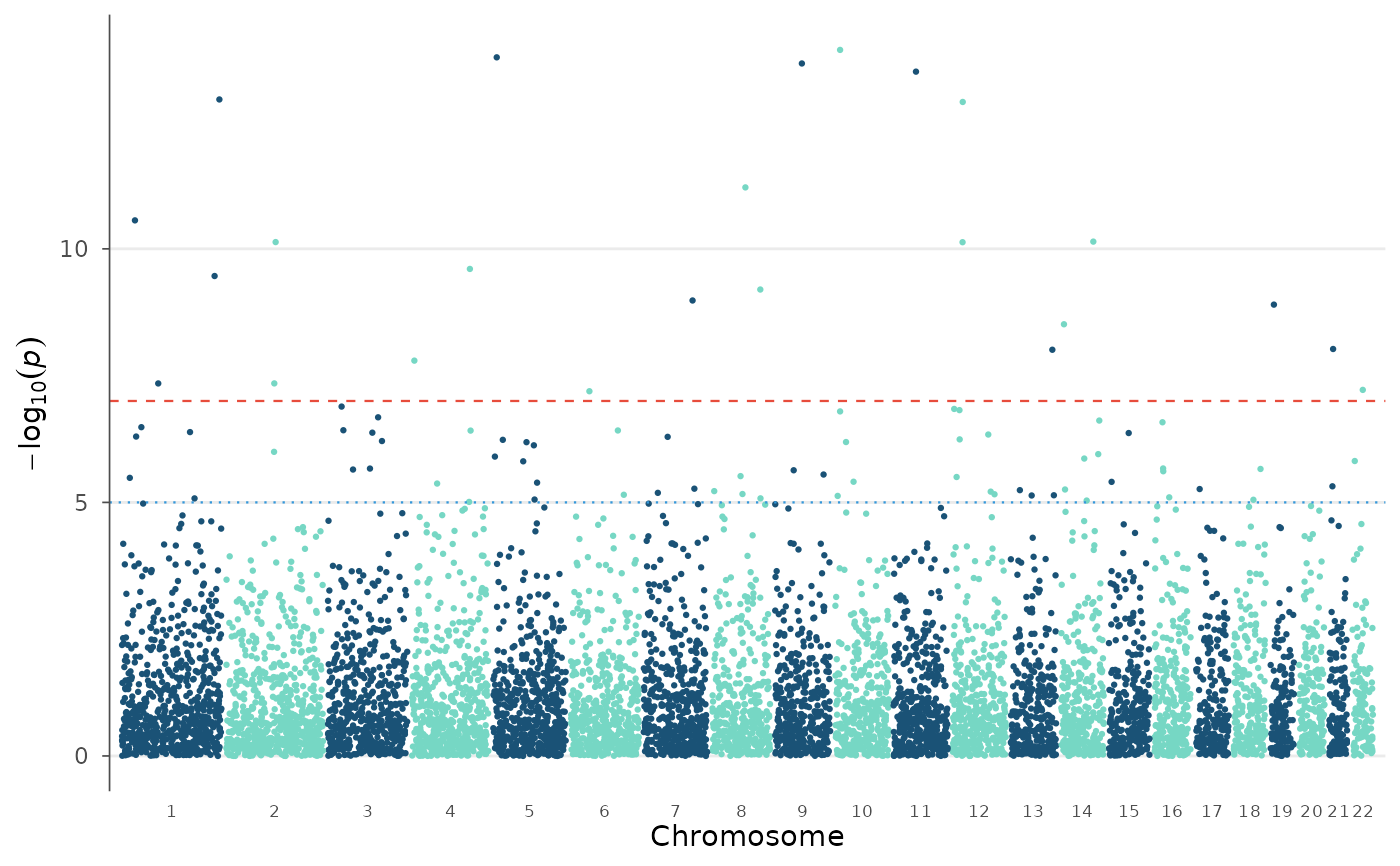 # No threshold lines
manhattan_plot(example_gwas, genome_wide = NULL, suggestive = NULL)
# No threshold lines
manhattan_plot(example_gwas, genome_wide = NULL, suggestive = NULL)
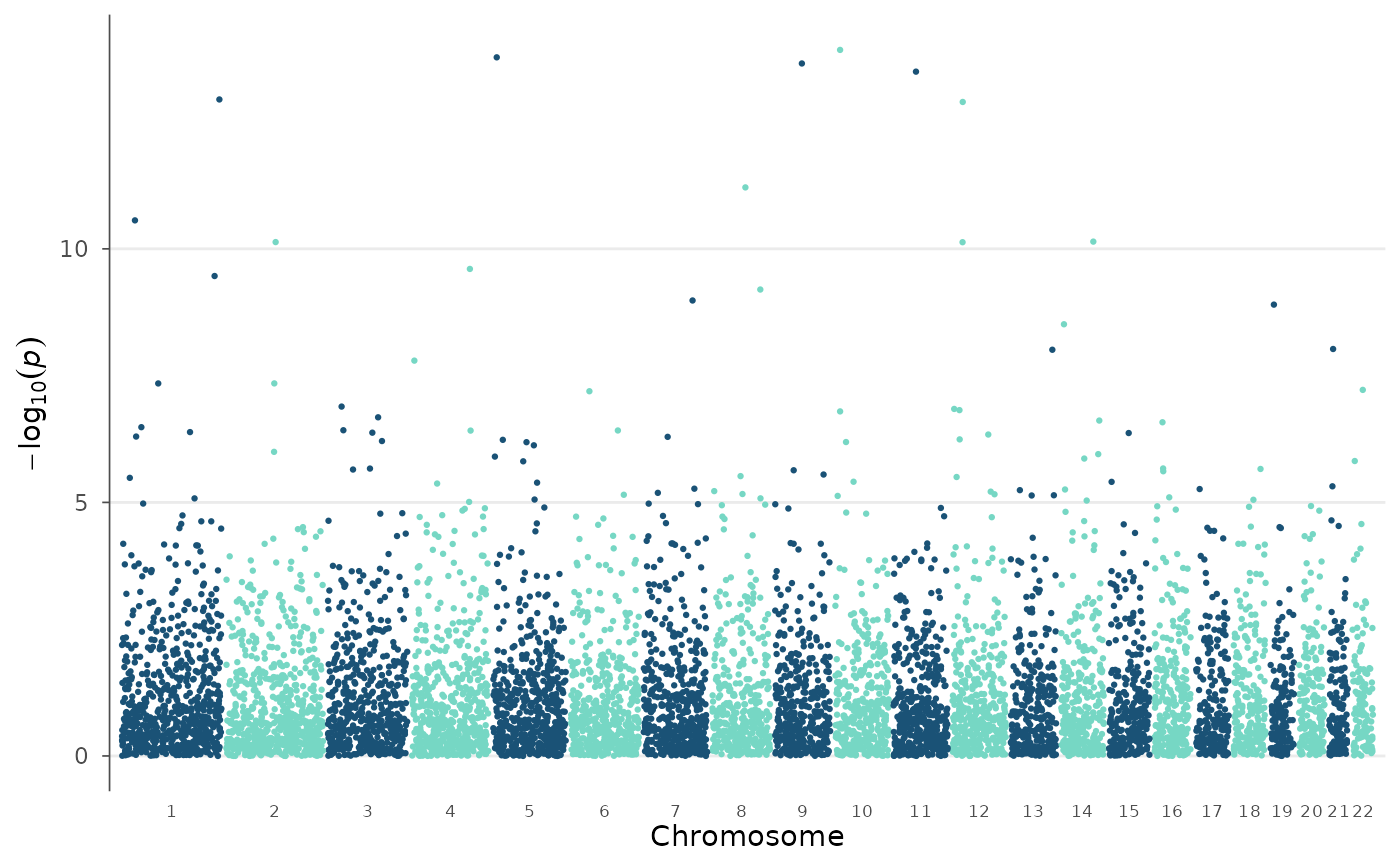 # Label only odd chromosomes (less crowded x-axis)
manhattan_plot(example_gwas, chr_labels = "odd")
# Label only odd chromosomes (less crowded x-axis)
manhattan_plot(example_gwas, chr_labels = "odd")
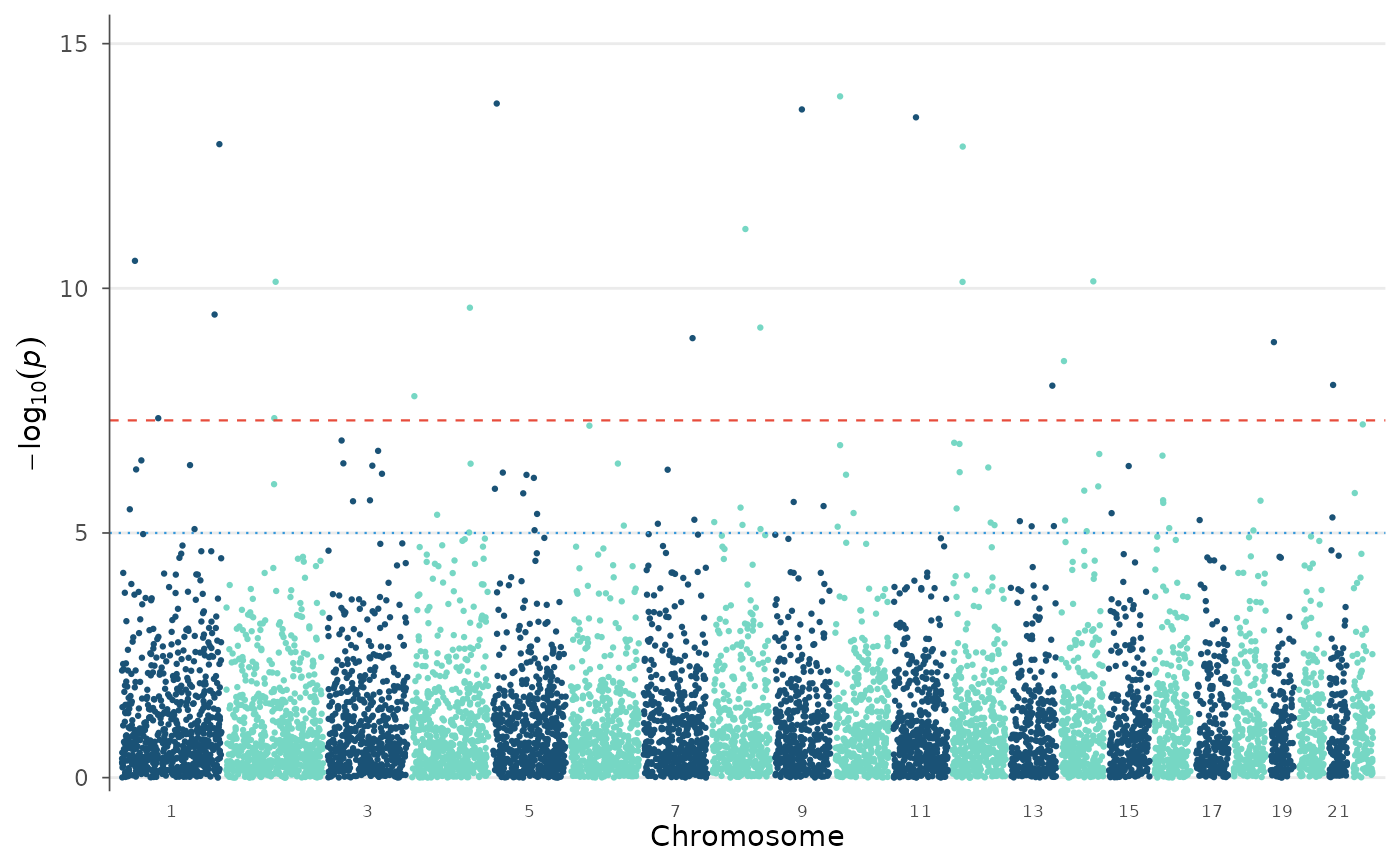 # Effect-size confidence bound (|beta| - 2*SE)
manhattan_plot(example_gwas, y_metric = "beta_min")
# Effect-size confidence bound (|beta| - 2*SE)
manhattan_plot(example_gwas, y_metric = "beta_min")