Fast, customizable GWAS visualizations built on ggplot2, with sensible defaults and journal-specific themes for publication-ready figures.
Key features
- 20 plot types: from classic Manhattan and QQ to post-GWAS visualizations (PheWAS, colocalization, fine-mapping, genetic correlations, SNP density, forest, effect comparison, and a power-contour trumpet plot)
- Built-in gene models (GRCh37/38) and exon-level gene tracks — regional plots without a GTF
- Genomic tracks: composable gene annotation panels from GTF/GFF3 files with strand arrows and highlighting
-
Broken y-axis for Manhattan plots with extreme p-values (
y_truncate) -
Effect-size confidence mode showing |beta| - 2*SE instead of p-values (
y_metric = "beta_min") - Smart downsampling for 10M+ variant datasets
- Journal themes (Nature, Science, Cell, PLOS) and 14 color palettes
- Gene annotation with automatic nearest-gene mapping
- Auto-detects column names from PLINK, REGENIE, GCTA, GEMMA, and generic files
- Fully composable: every function returns a ggplot object
Gallery
| Manhattan plot | Broken y-axis |
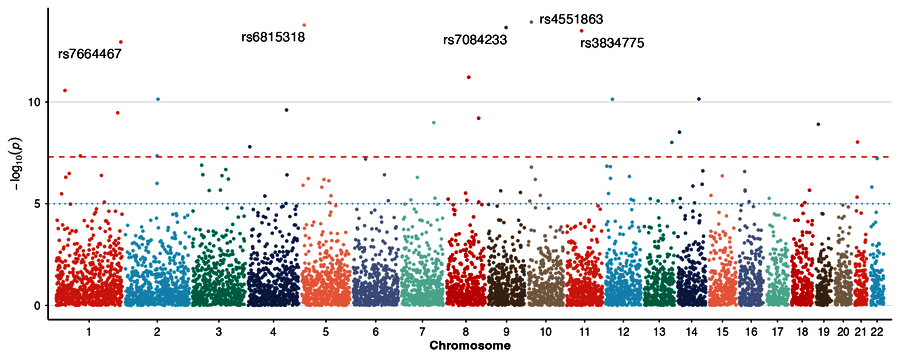 |
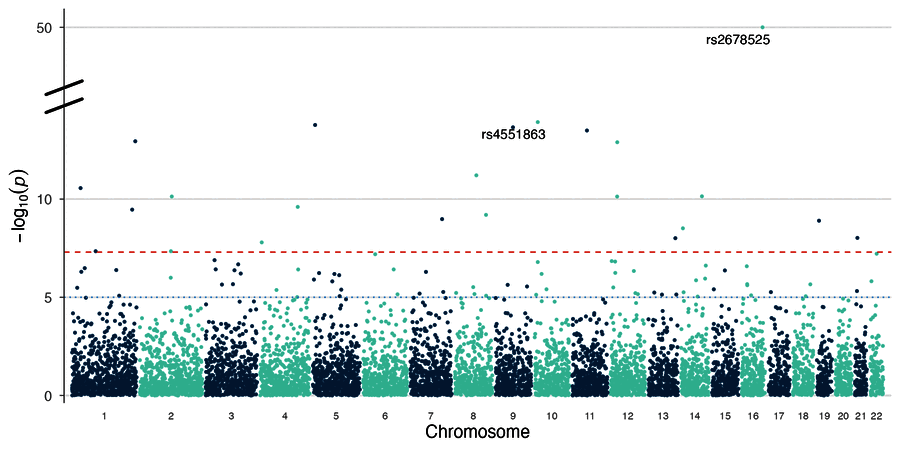 |
| QQ plot | Circular Manhattan |
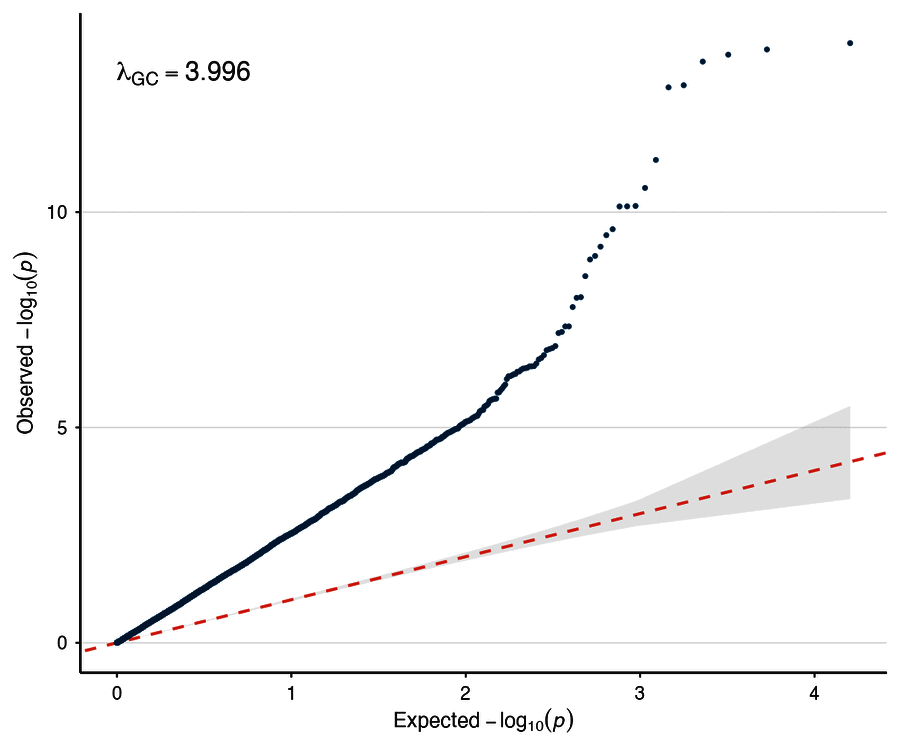 |
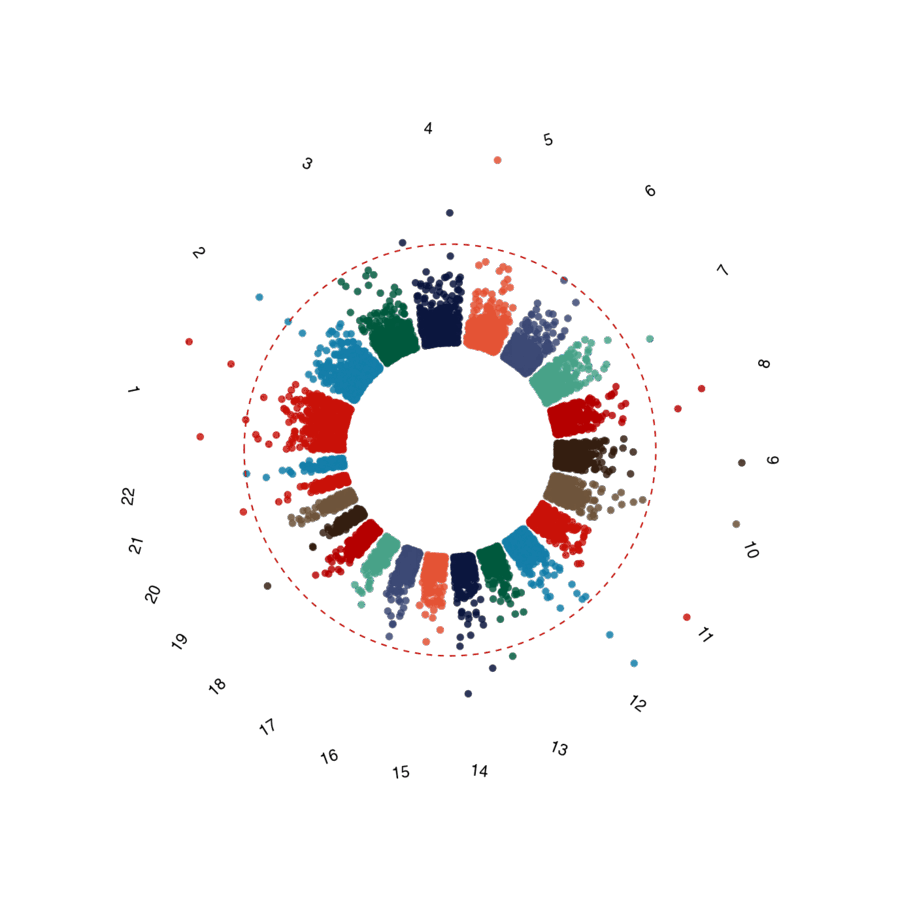 |
| Miami plot | Multi-trait Manhattan |
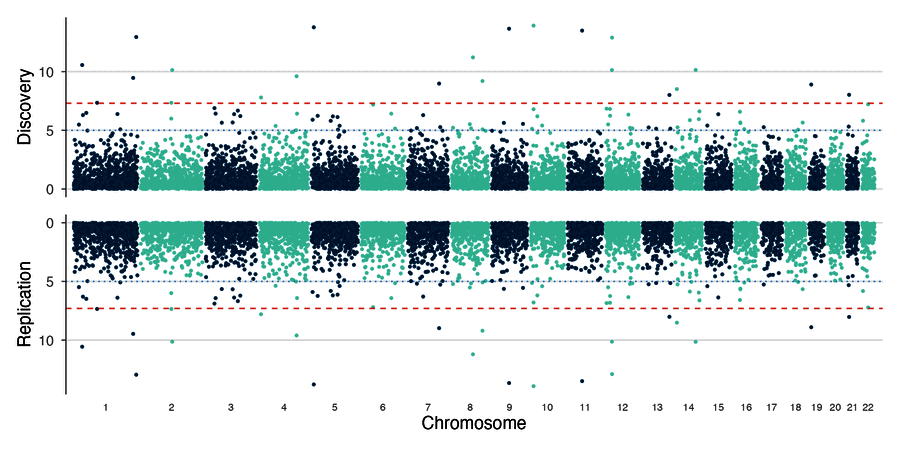 |
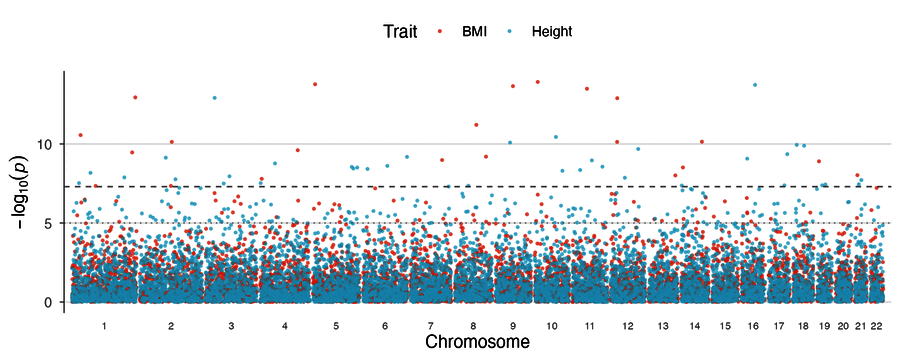 |
| Enrichment Manhattan | PheWAS plot |
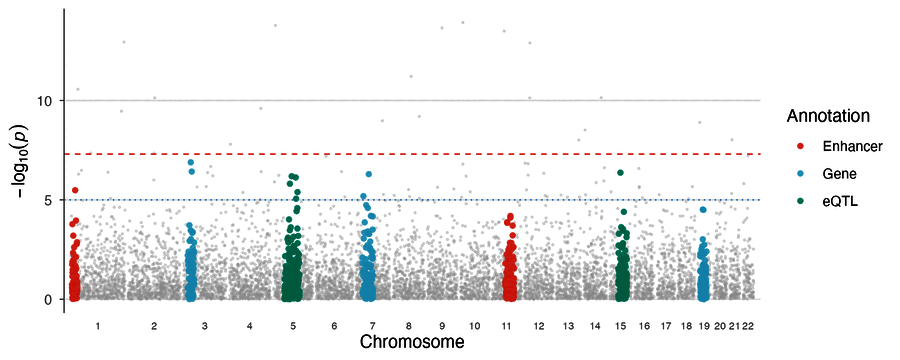 |
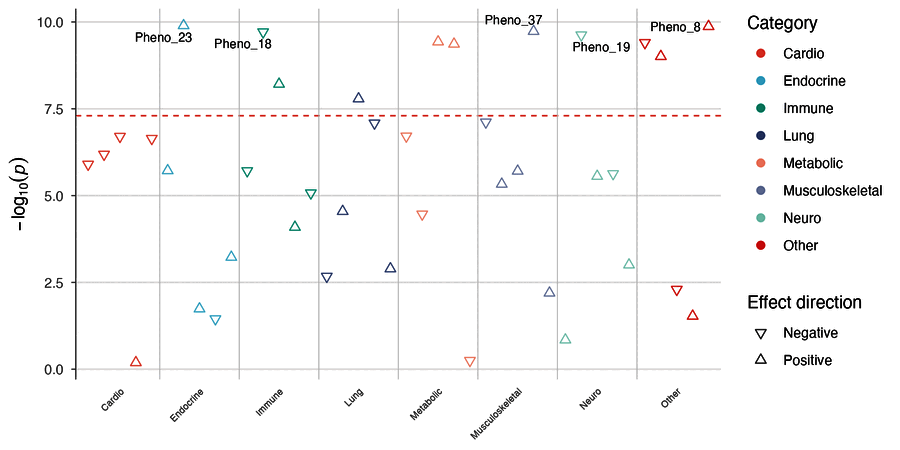 |
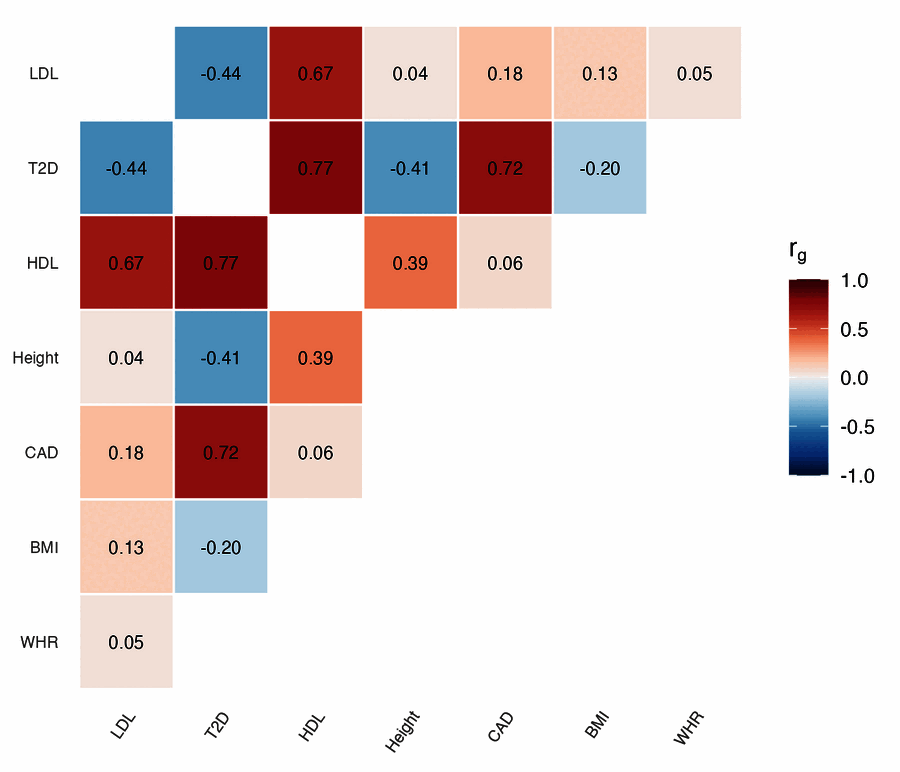
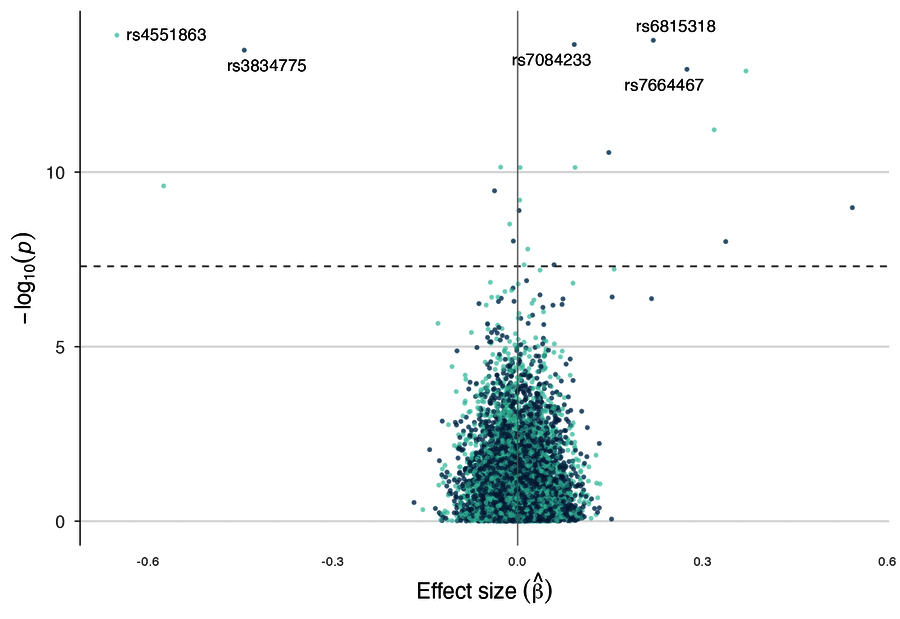
| Genetic correlation | Effect-size volcano |
 |
 |
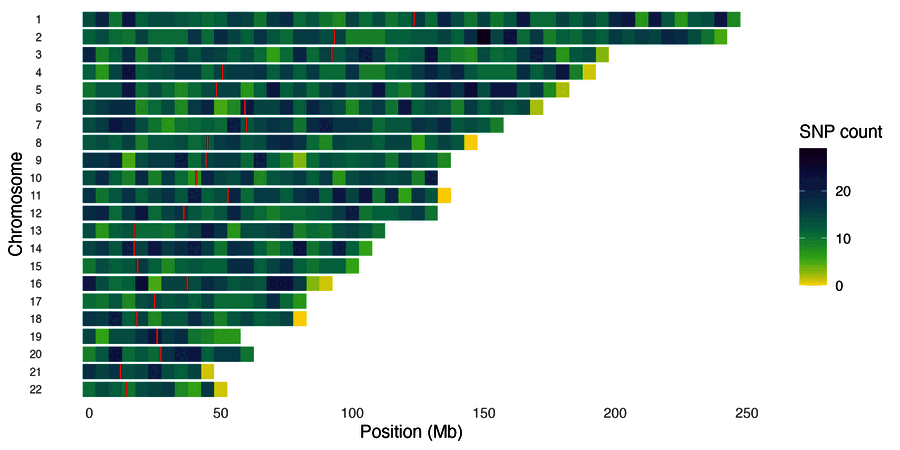
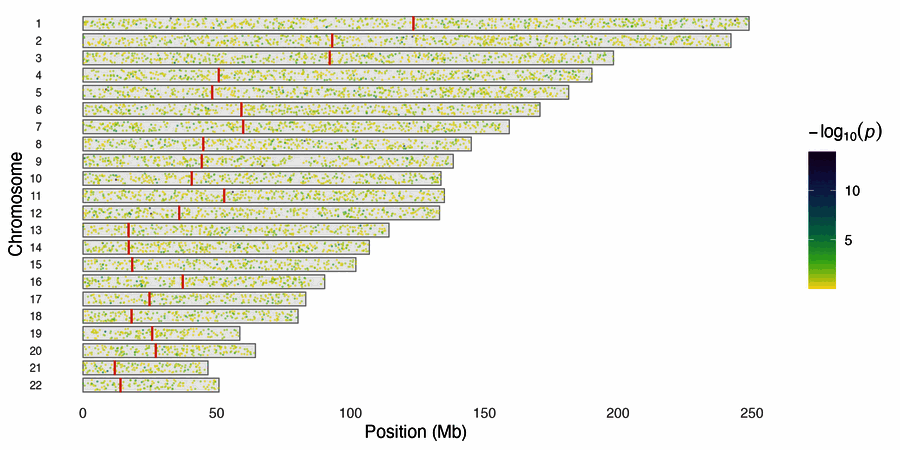
| SNP density karyogram | SNP density (points) |
 |
 |
| Density vs signal | P-value heatmap |
 |
 |
| Locus zoom | Effect-size confidence |
 |
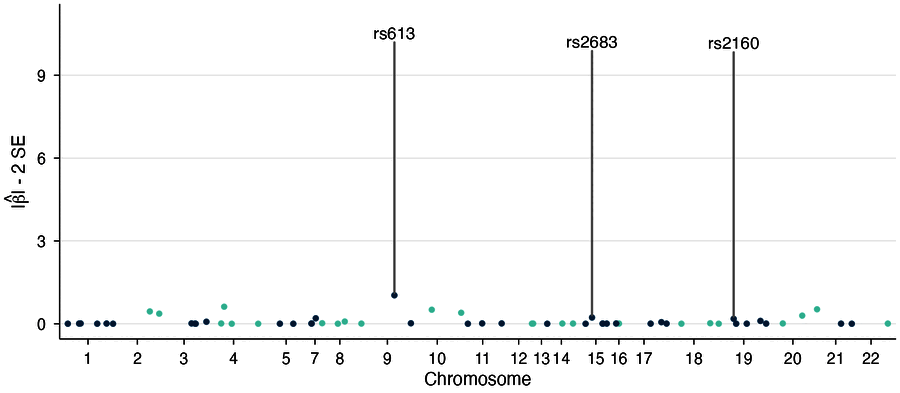 |
| Genetic architecture | Journal themes |
 |
 |
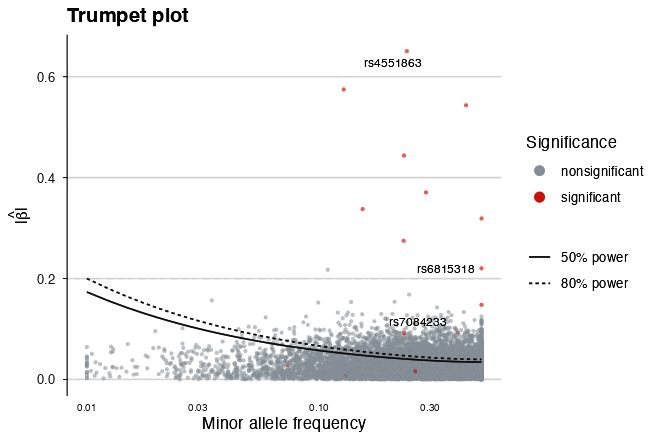
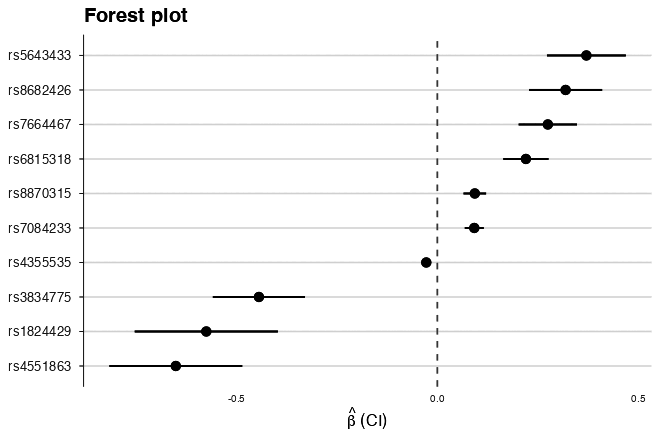
| Trumpet plot (power contours) | Forest plot |
 |
 |
| Effect comparison | Gene track with exons |
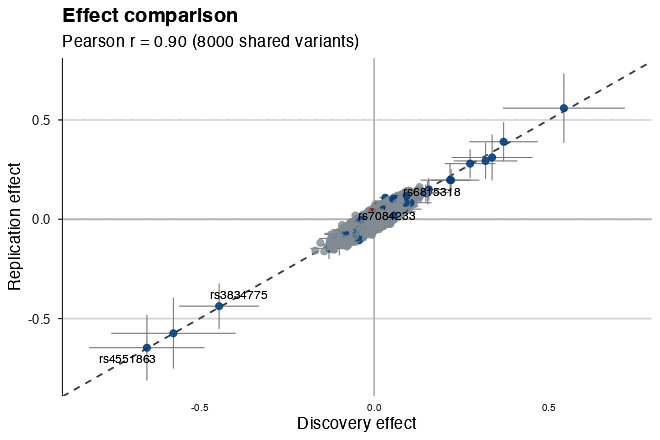 |
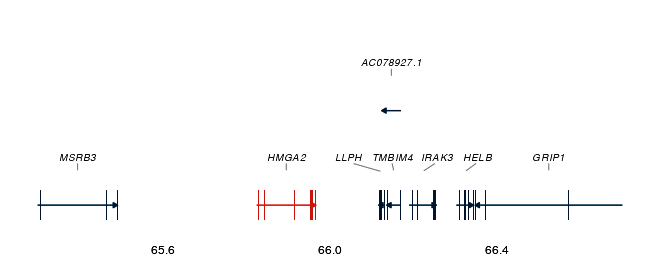 |
Full documentation with worked examples: https://bczech.github.io/ggwas/
Comparison
The closest ggplot2-native package is topr; ggwas matches its regional/gene depth and adds the genome-wide and post-GWAS breadth below.
| Feature | qqman | CMplot | topr | ggwas |
|---|---|---|---|---|
| ggplot2-native | No | No | Yes | Yes |
| Manhattan + QQ | Yes | Yes | Yes | Yes + CI + λ + MAF-stratified |
| Miami plot | No | No | No | Yes |
| Locus zoom | No | No | Yes | Yes (LD + gene track) |
| Circular Manhattan | No | Yes | No | Yes (multi-ring) |
| Enrichment Manhattan | No | No | No | Yes |
| Multi-trait overlay | No | No | Yes | Yes (pleiotropy) |
| Genome-wide heatmap | No | No | No | Yes |
| Effect-size volcano | No | No | No | Yes |
| Summary dashboard | No | No | No | Yes |
| PheWAS plot | No | No | No | Yes |
| Colocalization | No | No | No | Yes |
| Fine-mapping (PIP) | No | No | No | Yes |
| Genetic correlation | No | No | No | Yes |
| Architecture plot | No | No | No | Yes |
| Trumpet (power) plot | No | No | No | Yes |
| Forest plot | No | No | Yes | Yes |
| Effect comparison | No | No | Yes | Yes |
| SNP density karyogram | No | Yes | No | Yes |
| Density vs signal | No | No | No | Yes |
| Gene track (with exons) | No | No | Yes | Yes |
| Built-in gene models | No | No | Yes | Yes (GRCh37/38) |
| GRanges interoperability | No | No | No | Yes |
| Region highlights | No | No | No | Yes |
| Top hits (clumping) | No | No | No | Yes |
| Journal themes | No | No | No | 6 themes + 4 presets |
| Color palettes | Limited | Limited | Limited | 14 (colorblind-safe) |
| Auto-detect formats | No | No | No | Yes |
| Downsampling | No | No | Yes | Yes |
Installation
pak::pak("bczech/ggwas")Quick start
library(ggwas)
# Try it on the bundled example dataset
data(example_gwas) # a data.frame of GWAS results (CHR, BP, SNP, P, ...)
manhattan_plot(example_gwas)
# Read any GWAS results file (columns auto-detected)
gwas <- read_gwas_table("my_results.txt")
# Manhattan plot
manhattan_plot(gwas)
# Label top hits with gene names
manhattan_genes(gwas, genes = my_gene_table, gene_top_n = 10)
# QQ plot with confidence band and lambda
qq_plot(gwas, show_lambda = TRUE)
# Miami plot: discovery vs replication
miami_plot(discovery, replication,
top_title = "Discovery", bottom_title = "Replication")
# Publication preset
p <- gwas_preset("publication")
manhattan_plot(gwas, colors = p$colors, point_size = p$point_size) + p$themeSupported input formats
| Format | Function |
|---|---|
| Generic (auto-detect) | read_gwas_table() |
| PLINK .assoc/.linear/.logistic |
read_plink_assoc() / _linear() / _logistic()
|
| REGENIE | read_regenie() |
| GCTA MLMA | read_gcta_mlma() |
| GEMMA | read_gemma() |
| Any data.frame | Pass directly with column mapping |
Plot gallery
Core plots
manhattan_plot(gwas, label_top_n = 5)
manhattan_plot(gwas, y_truncate = 15) # broken y-axis for extreme p-values
manhattan_plot(gwas, y_metric = "beta_min") # effect-size confidence bound
qq_plot(gwas, show_lambda = TRUE, ci = 0.95)
miami_plot(gwas1, gwas2, top_title = "Study 1", bottom_title = "Study 2")
locus_plot(gwas, lead_snp = "rs12345", flank = 500000)Novel visualizations
pvalue_heatmap(gwas, bin_size = 1e6, palette = "magma")
volcano_plot(gwas, label_top_n = 10, color_by = "chromosome")
circular_manhattan(gwas, colors = gwas_palette("nature"))
circular_manhattan(list(BMI = gwas1, Height = gwas2)) # multi-ring
enrichment_manhattan(gwas, annotations = functional_regions)
multitrait_manhattan(BMI = gwas1, Height = gwas2, highlight_shared = TRUE)
trumpet_plot(gwas, n = 500000) # effect vs MAF with statistical-power contours
gwas_summary(gwas) # multi-panel dashboardPost-GWAS
phewas_plot(phewas_results)
coloc_plot(gwas, eqtl, region_chr = 1, region_start = 1e6, region_end = 2e6)
finemapping_plot(susie_results, region_chr = 1, region_start = 1e6, region_end = 2e6)
genetic_correlation(ldsc_matrix, cluster = TRUE)
architecture_plot(gwas)
snp_density(gwas, chr_info = chr_info_human())
density_signal_plot(gwas)
forest_plot(lead_variants) # effect estimates + CI
effect_compare_plot(discovery, replication) # beta-vs-beta concordanceGene annotation and genomic tracks
# Built-in gene models (GRCh37/38) — no GTF or biomaRt needed
genes <- gene_annotation("GRCh38")
# ...or read your own: genes <- read_gtf("Homo_sapiens.GRCh38.110.gtf.gz")
# Gene track below a locus plot (add exon_data for exon structure)
p <- locus_plot(gwas, region_chr = 6, region_start = 25e6, region_end = 35e6)
gt <- gene_track(genes, region_chr = 6, region_start = 25e6, region_end = 35e6,
highlight_genes = c("HLA-A", "HLA-B"))
patchwork::wrap_plots(p, gt, ncol = 1, heights = c(0.8, 0.2))
# Label peaks with nearest gene names (instead of rs IDs)
manhattan_genes(gwas, genes = gene_table, arrow = TRUE)
# Extract independent top hits with gene mapping
top_hits(gwas, genes = gene_table, p_threshold = 5e-8)
# Highlight genomic regions
plt <- manhattan_plot(gwas)
highlight_regions(plt, data.frame(chr = 6, start = 25e6, end = 34e6, label = "MHC"))Themes and palettes
# Journal themes
manhattan_plot(gwas) + theme_nature()
manhattan_plot(gwas) + theme_science()
manhattan_plot(gwas) + theme_cell()
manhattan_plot(gwas) + theme_plos()
# Presentation / poster
manhattan_plot(gwas) + theme_presentation()
manhattan_plot(gwas) + theme_poster()
# 14 color palettes
gwas_palettes()
manhattan_plot(gwas, colors = gwas_palette("nature"))Performance
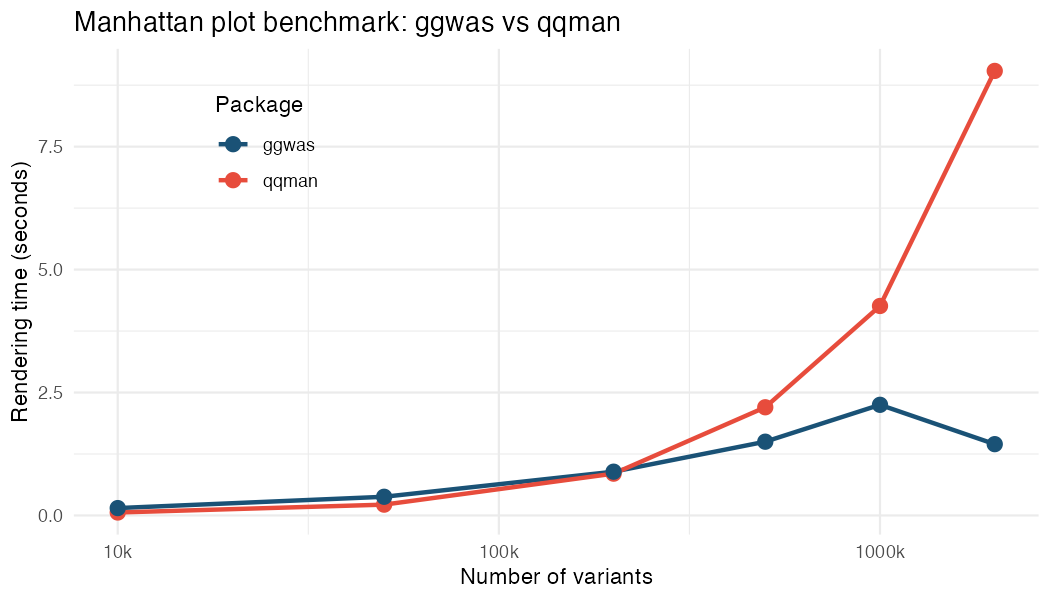
Smart downsampling keeps every significant variant and spatially bins the denser background, capping the number of plotted points. Render time therefore stays roughly flat as datasets grow, while base-R (qqman, CMplot) and other ggplot2 tools (topr) scale with variant count. Median of 5 runs on the GIANT height GWAS, Manhattan render time in seconds:
| Variants | qqman | CMplot | topr | ggwas |
|---|---|---|---|---|
| 50k | 0.33 | 0.21 | 0.26 | 0.19 |
| 200k | 1.31 | 0.70 | 0.73 | 0.53 |
| 500k | 3.12 | 1.65 | 2.01 | 1.26 |
| 1M | 6.56 | 3.37 | 3.96 | 1.48 |
| 1.37M | 9.15 | 6.32 | 6.61 | 1.30 |
manhattan_plot(large_gwas) # 10M SNPs, auto-downsampled to ~200k pointsDocumentation
Full documentation with worked examples: https://bczech.github.io/ggwas/
Or from R:
vignette("ggwas")